
一、适用范围
该指南适用于以下三种情形的抗癌药物联合疗法的开发:
• 两种(或多种)尚未获得FDA任何适应症批准的研究性药物联合使用;
• 一种研究性药物与已获批其他适应症的药物联合使用;
• 两种(或多种)已获批其他适应症的药物联合使用。
但不适用于以下情况:
• 研究性药物与已获批相同适应症的药物联合开发(即标准治疗的加载研究,add-on study);
• 将先前批准药物开发为固定组合制剂用于相同适应症。
此外,该指南不涉及联合用药试验中的安全性或剂量设计考量,不涉及对安全性数据进行评估以支持联合治疗的风险-获益评估。
二、背景
肿瘤联合疗法作为一种重要的治疗模式,其发展得益于科学进展对肿瘤病理生理机制的深入解析。这种认知深化进一步推动了多靶点联合用药策略的开发,以提升疗效、降低不良反应,或二者兼得。当组合中每个药物的必要性均具备有力的生物学依据(包括组合内各药物的非临床特性及早期临床证据)时,该新药组合可进入开发阶段。此类依据包括但不限于:
• 药物分别抑制同一分子通路中的不同靶点,或干预发病机制中的不同环节,实现对核心通路或代偿通路的双重抑制;或在同一靶点的不同结合域发挥作用,以降低耐药性或减少给药剂量从而控制整体毒性。
• 药物单用时疗效有限,但基于相关非临床研究/短期临床生物标志物研究,证实其可增强组合中另一药物的协同治疗效应。
• 早期临床试验显示单药疗效不足,但联合用药可能产生显著治疗优势,成为更优治疗选择。
三、证明抗癌药物在联合疗法中的疗效贡献
对于创新联合疗法的新药上市申请,必须确证整体的安全性和有效性,并提供充分证据证明各组分对疗效具有实质性贡献。FDA鼓励开发肿瘤新型联合疗法的申办方尽早(如通过Pre-IND会议),并在临床开发期间与FDA审评部门保持沟通,尤其对复杂开发项目,以获取FDA对可接受开发路径的反馈,并可能有助于建立简化开发策略。
支持评估每种药物疗效贡献所需的临床数据和试验设计的数量和类型可能各不相同,取决于疾病和人群的背景、其他治疗的可用性和有效性、可用的临床前和临床数据、单个药物和联合的临床数据范围,以及正在研究的问题的复杂性。若条件允许,建议采用析因设计随机试验(Factorial Design Randomized Trial),以提供足够的证据来证明单个药物对新型联合治疗的疗效贡献。
A. 证明疗效贡献的析因设计(Factorial Design)
抗癌药物联合疗法的传统评估通常采用多臂随机对照试验设计,包括联合用药组、各单药治疗组、和非单药标准治疗组(见下图)。这种析因设计不仅可以对联合用药及单药相较于标准治疗的安全性和有效性进行评估,还可以展示单药对联合治疗整体疗效的贡献。2013年《联合开发指南》第V.C节讨论了多种用于评估研究药物疗效贡献的试验设计,适用于不同情境,如:药物可单独给药且各自有活性、不可单独使用,或其中一种疗效有限等。
当联合治疗中每种药物均具活性时,建议采用随机化析因设计,以明确各组分对整体疗效的贡献。
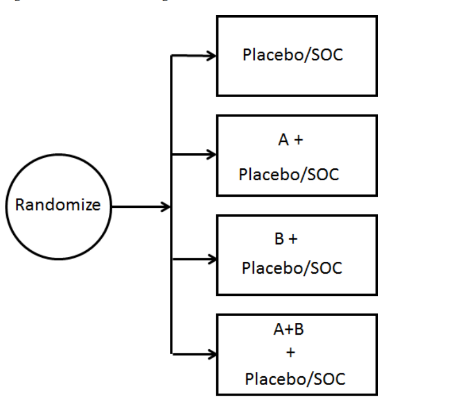
图:析因试验设计 (A: 单药A;B: 单药B;SOC: 标准治疗)
在联合用药方案中,如果已有证据表明某研究药物单用无效,可采用三臂的改良析因设计(modified factorial design)以明确各组分对联合疗效的贡献。适应性析因设计(adaptive factorial design)可提高新型联合用药的开发效率,此设计在初始阶段将受试者随机分至各试验组,试验过程中可终止可能无效的实验组或作其他调整,从而减少总体样本量及暴露于低效方案的风险。
旨在证明各组分的疗效贡献的试验,其主要终点可选择具说服力的药效学或反应性生物标志物,能够直接提供某种癌症治疗效果的证据(如总体缓解率及缓解持续时间),且相较其他关键临床终点(如无进展生存期或总生存期)具备更早可测性优势。但其局限性在于:部分组分可能对长期终点(如无进展生存期或总生存期)未产生实质贡献或存在不利影响。因此,整体开发计划应确保各单一成分对疗效确有贡献。FDA鼓励申办方与其沟通,明确试验设计框架与终点选择。
B. 证明疗效贡献的外部数据
使用外部数据支持单药在联合疗法中疗效贡献的前提包括:1)该联合治疗方案具有充分的生物学合理性;2)该疾病的自然病程高度可预测;3)有证据表明该药物单独使用时的疗效低于其与其他类别药物联合使用时的疗效;和/或 4)预期的联合治疗方案疗效非常显著。在考虑采用此方法时(以及其他相关因素),所用外部数据应满足以下条件:• 与目标人群具可比性;
• 包含对临床相关的混杂变量的详细信息;
• 疗效评估方法一致。
此方法的挑战在于选择合适的终点指标。建议在使用外部数据支持疗效贡献前,先与FDA沟通研究方案。
1. 外部数据来源在疗效贡献评估中的适用性
数据来源的适用性由多个与试验各组可比性相关的因素决定,这些因素可能包括但不限于:各组分治疗效果贡献的时间关联性、受试者来源人群、纳入和排除标准(包括疾病类型及治疗线别)的可用性和相似性、以及试验参与者数据的可获取性。申办方应收集充分的受试者人口学和临床特征信息,重点关注预后因素和预测因素的平衡,以充分评估不同数据来源之间的可比性,确保疗效评估的内部有效性。
申办方在使用外部数据前应评估如下关键因素:
• 疾病自然病程的已知信息;
• 标准治疗背景下的结局数据(如肿瘤缓解率),特别是当SOC为联合方案的一部分时;
• 患者级数据可用性,包括足够的样本量和随访时间以支持数据源可比性分析;
• 关键数据的可追溯性、可审计性,确保数据与准确、可靠、有效;
• 缺失数据的程度,包括暴露信息和关键协变量的缺失情况;
• 维持盲态筛选参与者的可行性;
• 预设明确的统计分析计划,用于评估各组分的疗效贡献。
如果外部数据在人群特征、研究方式或终点评估方面与试验存在差异,可考虑使用因果推断、贝叶斯等方法进行调整,但这些方法不能完全消除所有偏倚,结果解读仍应保持谨慎。
2. 外部数据源的选择
• 同期临床试验数据(相同背景及适应症):尤其在与本研究时间重叠、数据具有时效性的情况下,可提供高相关性证据;而历史数据可能会引入时间偏倚。
• 前瞻性收集的患者水平数据(如登记库数据):需包括人口统计学信息、疾病特征、治疗情况及相关结局等。
• 其他患者级别的真实世界数据来源。
• 既往已发表临床试验或观察性(非干预性)研究的汇总级别证据。
3. 使用外部数据时的终点评估考量
• 时间事件终点:不仅基线测量可能受影响,事件时间的测量也可能引入偏倚(如永恒时间或生存期偏倚),需要验证指标日期与随访日期在各组间可比;
• 总生存期:虽定义明确,但某些真实世界数据来源中死亡数据不全,且受后续抗癌治疗的介入影响较大;
• 肿瘤负荷终点(如缓解率/进展期):需确保评估频率与评价标准一致;
• 其他终点:经验证的患者报告结局、临床结局评估指标、或生物标志物可作为补充支持。
C. 证明疗效贡献的开发策略考量
1. 两个或多个在研药物
2. 一种在研药物与一种已在其他适应症中获批的药物联合
• 在研药物尚无任何适应症下的安全性或有效性证据;
• 目标适应症自然病程复杂,难以识别疗效;
• 联合疗法总体疗效幅度有限。
若具有充分的生物学机制支持以及来自非临床或早期临床的证据,能够降低外部数据比较带来的不确定性。在此类情况下,使用来自以往临床试验的外部数据,特别是针对已批准药物单药治疗该适应症的临床试验数据,可作为支持该组分在联合疗法中疗效贡献的依据,但仍需谨慎评估其相关性和可靠性。
总体上,鉴于外部数据的局限性,FDA 更倾向于在开发早期采用随机对照设计,并以总缓解率或其他能直接反映抗肿瘤效应的终点来评估单药对组合疗效的贡献。
3. 两种或以上已分别获批用于不同适应症的药物
• 不同适应症之间在病因学方面的相似性(例如:肿瘤中的分子异常是否一致)或疾病的临床背景相似性;
• 基于药物作用机制的合理性是否充分,是否具有支持该联合用药在特定疾病中使用的科学依据;
• 外部数据的证据强度:包括数据来源的充分性、终点设置是否合适,是否能够合理体现各单药在其他适应症中对疗效的实际贡献;
• 是否有足量的临床数据支持单药在多个疾病类型中对联合疗效的作用(例如:已在多种肿瘤中显示出贡献);
• 该新型联合治疗方案所展现的临床获益的重要性(如是否对总生存期有显著提升)。
撰稿人:Zihan Wang, Danila Xiang